Krithika NarayanaswamyAssistant ProfessorComputational Chemistry & Combustion group, Thermodynamics and Combustion Engineering Lab, Department of Mechanical Engineering, Indian Institute of Technology Madras, Chennai - 600036 Email: krithika (at) iitm (dot) ac (dot) in Research Experience Faculty page Scholar page ResearchGate page Scholar Profiles @ IITM |

|
 ORCID Page
ORCID Page | My research focuses on development of chemical kinetic models to describe oxidation of fuels. I am interested in predicting global combustion characteristics of conventional and alternative fuels and interpreting these observations based on insights gained from molecular level kinetic descriptions. If you are interested in being a part of our team, peruse the list of available projects to identify your interests and write to me with a subject line that includes the title of the topic. Please note that I am *not* interested in hiring interns. |
|
Research highlights
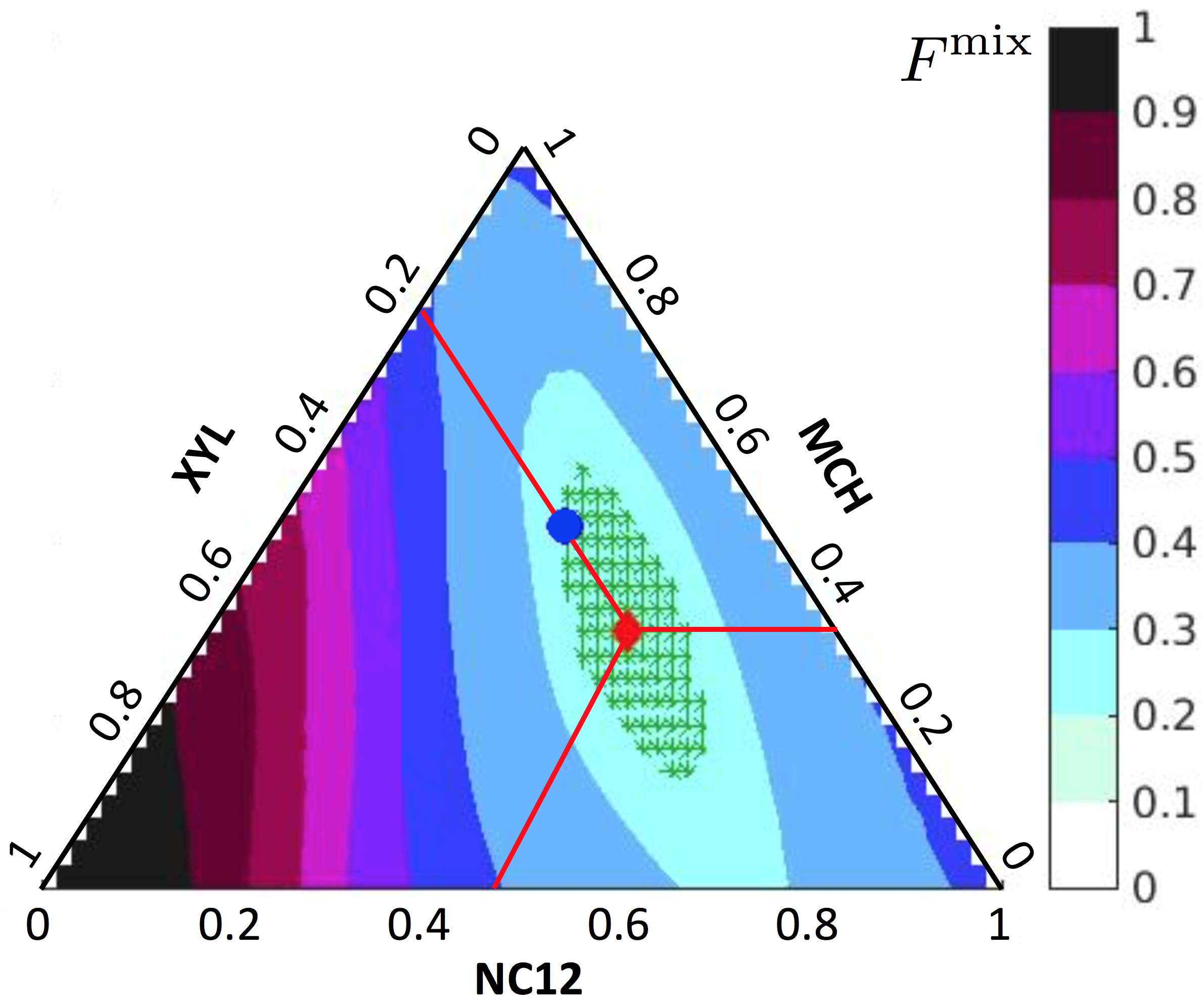
Simulation-driven surrogate formulation.
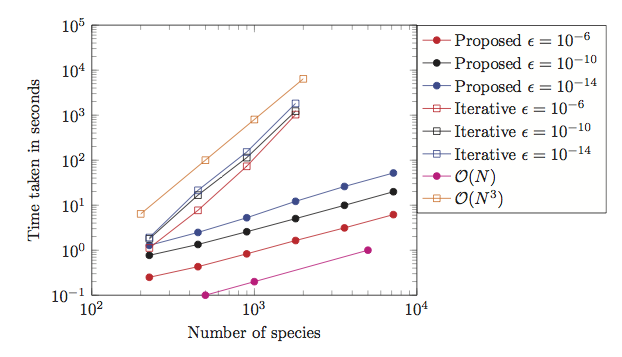
Time taken by our proposed fast algorithm to compute mulit-component diffusion velocities versus existing method.
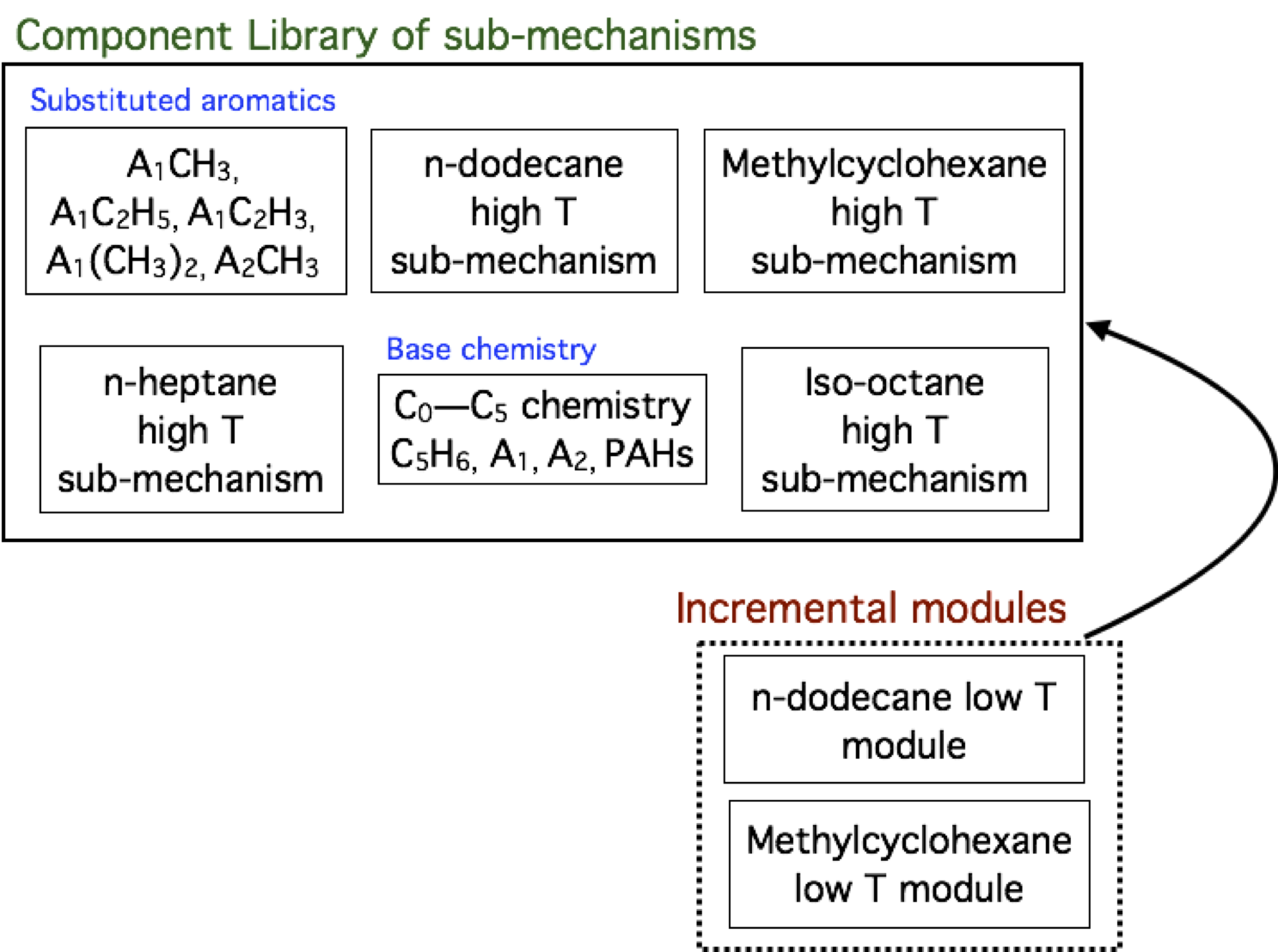
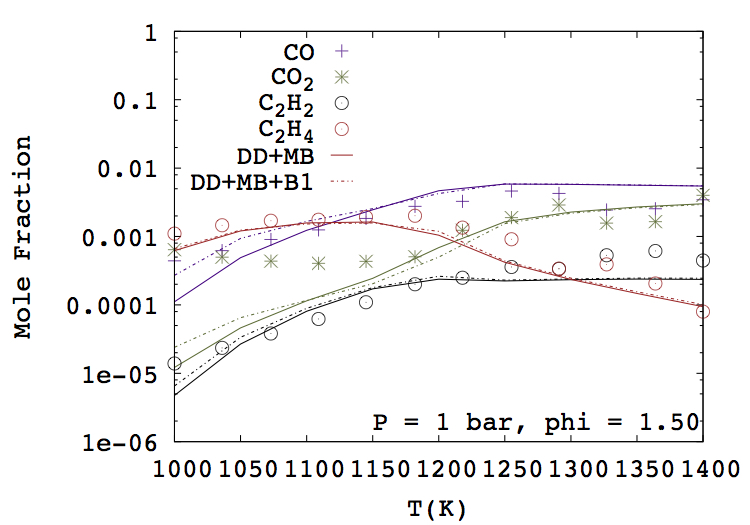
Chemical model developed in our earlier works reorganized into the component library framework.
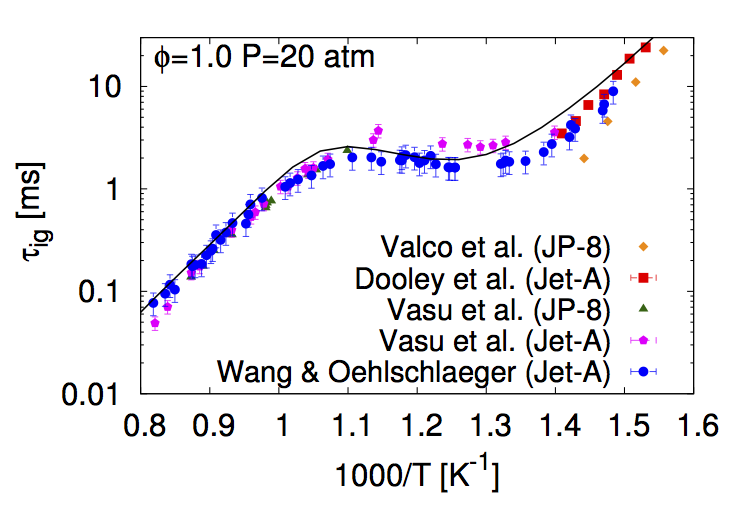
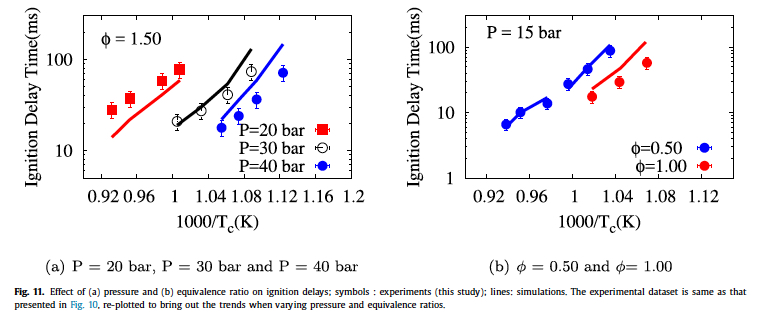
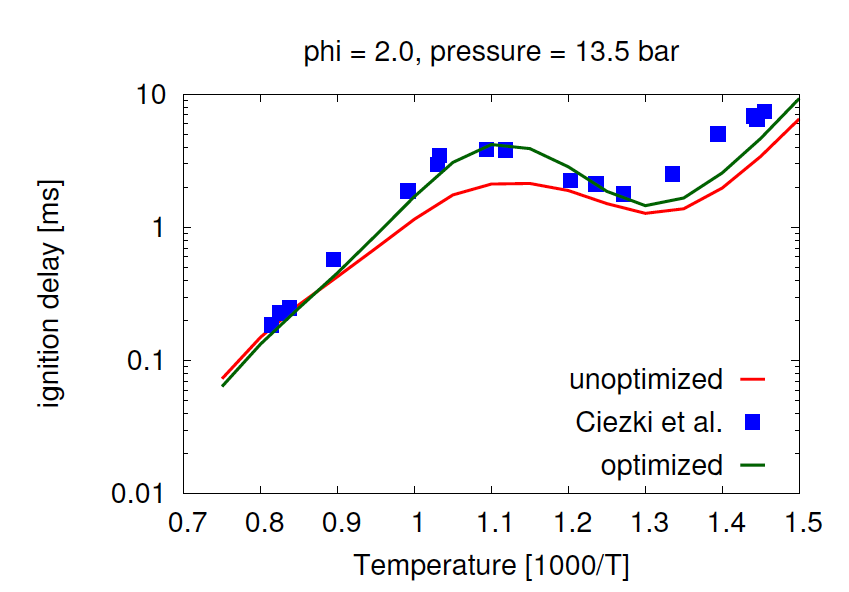
Ignition delays of jet fuel surrogate.
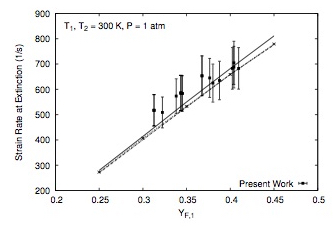
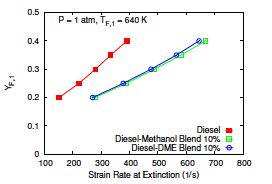
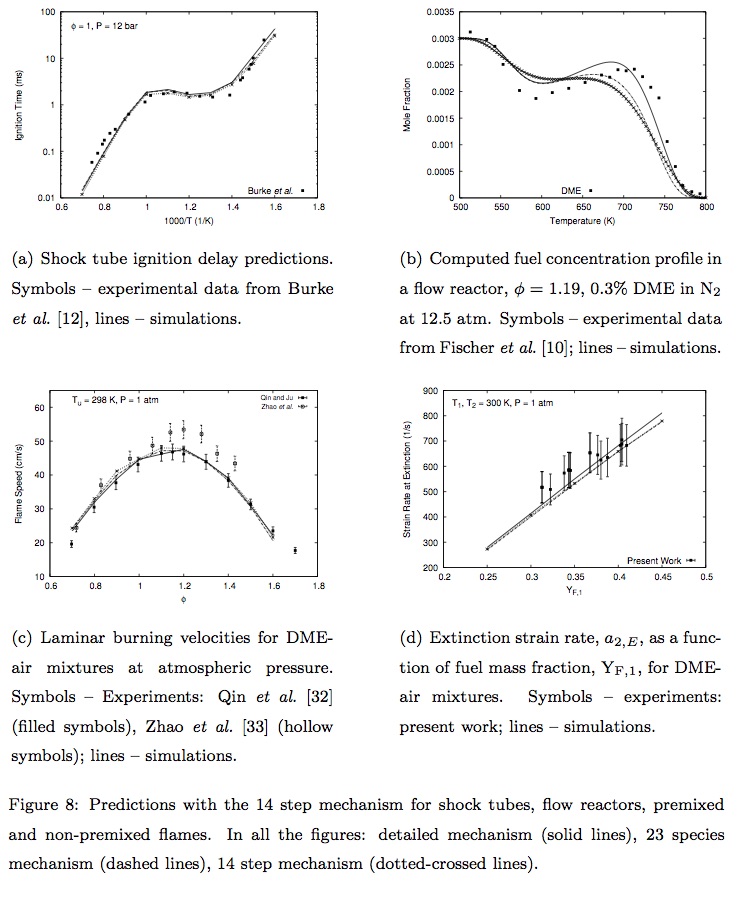
Extinction strain rates of DME-air mixtures measured in a counter-flow diffusion flame burner.
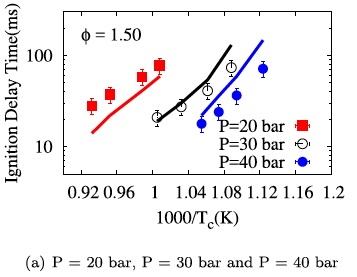
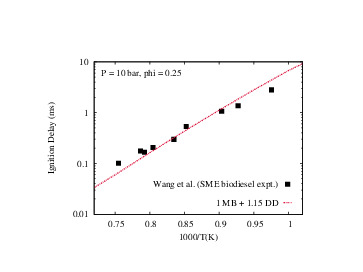
Ignition delay times of methylbutanoate.
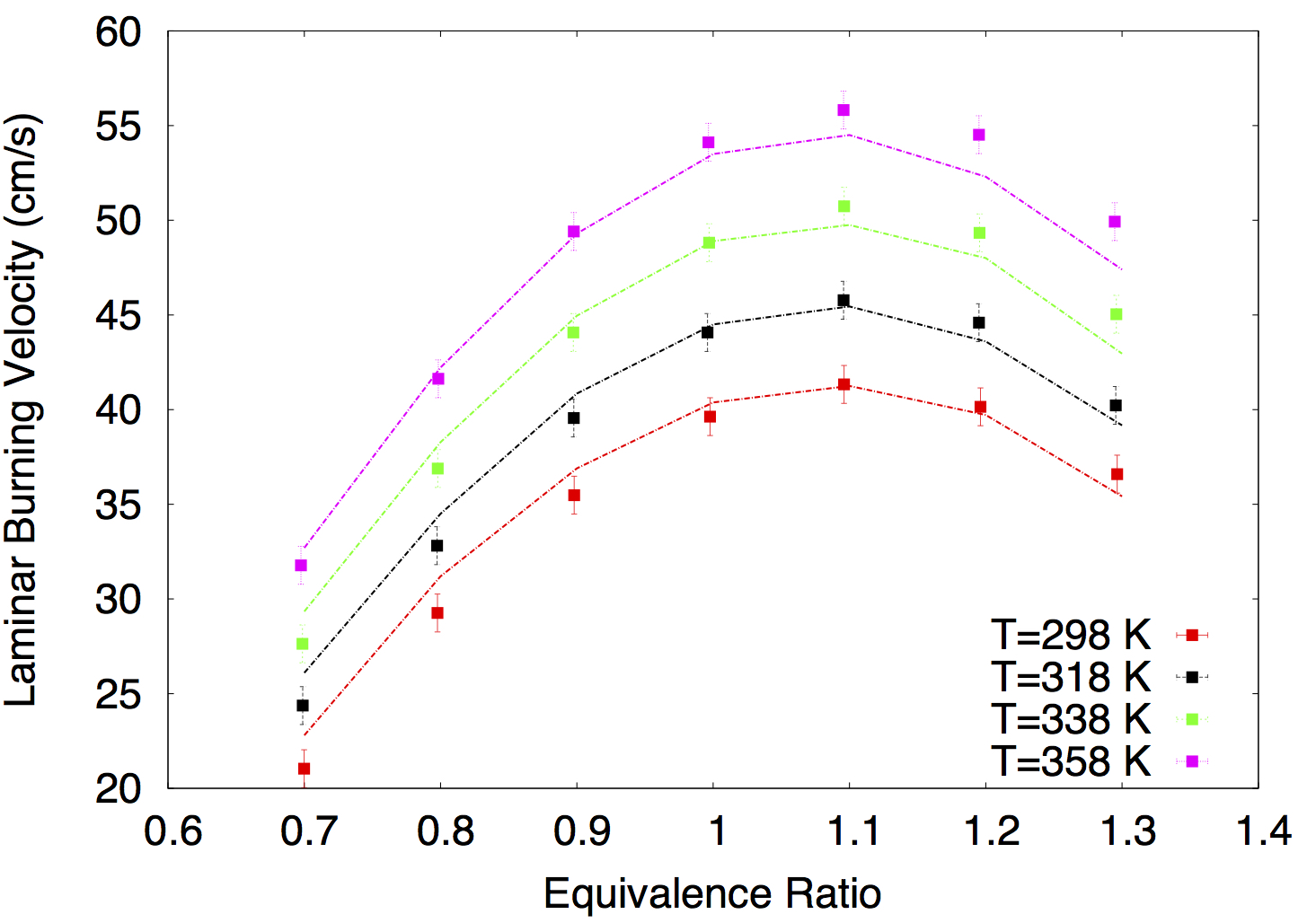
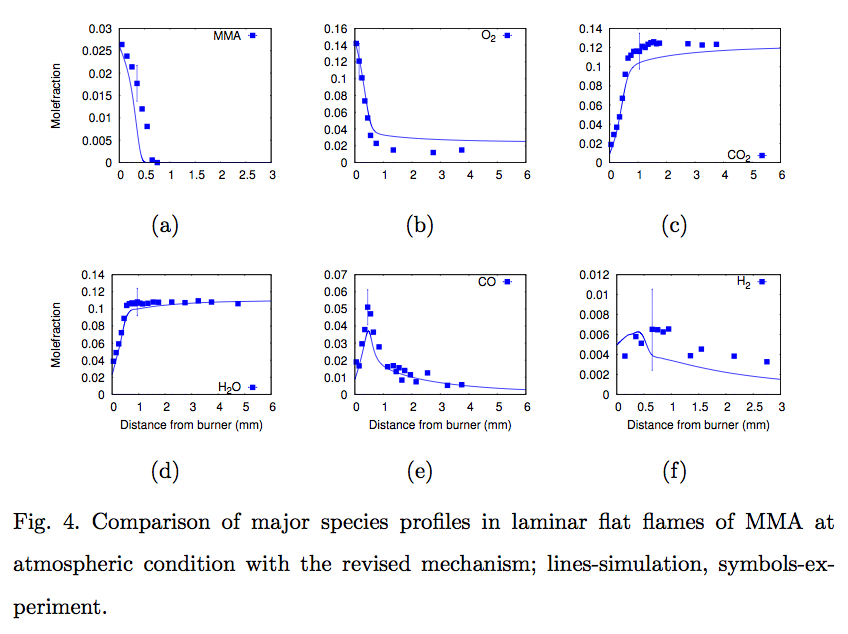
Flame speeds of methylmethacrylate at various unburnt fuel-air temperatures